| Contents |
|
- Requirements
- What the Scipio script does
- Usage
- Output overview
- Description of keys
- Sequence coordinate conventions
- Overview of Scipio parameters as activity diagram
- Detailed diagram of the Scipio application flow
| Requirements | Back to top |
- Bioperl (download from bioperl.org )
- YAML Perl module (download from CPAN)
- BLAT binary (see instructions at UCSC)
- Genome of choice in fasta format (you can search diArk for sequenced eukaryotic genomes)
| What the Scipio script does | Back to top |
Scipio locates the regions coding for a query protein sequence in a DNA target sequence. It is a convenient tool for the determination of exact gene structures and reverse translations.
For the computation of initial protein-DNA spliced alignments, Scipio utilizes the program BLAT written by Jim Kent. The output of BLAT is then further processed and reformatted by the following steps:
- BLAT does not try to align codons that are split by introns. The script searches for missing codons, preferring those that are split at splice sites, and adds the nucleotides to the corresponding exons.
- For each query sequence, most BLAT hits are discarded and only (the) one best hit is displayed. Scipio searches for hits and is optimized to reconstruct hits with nearly 100% sequence identity.
- Scipio is able to reconstruct the (may be not so) rare cases of genes that are found on different targets. First, all BLAT hits are collected and sorted by score. Then non-overlapping hits are taken to form a collection of hits of the same query.
- Frameshifts that usually cause BLAT to split exons into multiple separate matches, are joined back into a single match.
- Scipio retrieves all the corresponding sequences and groups them together with the BLAT results to form the output described below.
| Usage | Back to top |
| scipio.pl [<options>] <target> <query> | |
| <target> is a DNA file | |
| <query> is a protein file | |
| both in FASTA format | |
| General options | |
| --blat_output = <filename> | name of BLAT output file (.psl) to read from. Without this option, Scipio would start BLAT to produce a temporary output file that is processed and then deleted. With this option, Scipio will use the corresponding file instead of starting a new BLAT run. (However, if the file does not exist, BLAT will be started to create it.) This option might be useful if BLAT has already run separately, for example in parallel on multiple target files. |
| --overwrite (old --force_new) | use this option together with --blat_output to force overwriting the BLAT output file with a new BLAT run. |
| --blat_only | only call BLAT, do not run Scipio. |
| --partial_target | ignore lines in BLAT output files referring to nonexistent target sequences (by default, this causes an error). This is only needed if you ran BLAT separately and subsequently use Scipio on a subset of the target sequences. |
| --verbose | show verbose information (including progress) |
| --keep_blat_output | use this option if you don't want the BLAT output files to be deleted (this option is switched on automatically if --blat_output is given). |
| --show_blatline | together with --verbose: print out the commandline used by Scipio to call BLAT. |
| Options controlling choice of hits | |
| --min_score = <value> (old --best_size) | minimal score (between 0 and 1) of a hit for a query, in order to appear in the output. The formula for the score is: matches minus mismatches, devided by query length. If a hit is composed of multiple partial hits, the minimal score applies to the best-scoring partial hit. (default: 0.3) |
| --min_identity = <value> | minimal identity between query and target sequence in every single BLAT hit to be processed by Scipio, in percent (default is 90). |
| --min_coverage = <value> | minimal portion of the respective query sequence that must be found in every single BLAT hit to be processed by Scipio, in percent (default is 60). In the case of partial hits, we consider only the part between the first and the last residue of the query, that is aligned to the target sequence, for calculating the coverage. Gaps at query start or end, or between partial hits have no influence on the coverage. |
| --max_mismatch = <value> | maximal number of mismatches in a hit. Use 0 to allow any number of mismatches (default is 0). |
| --multiple_results | if this option is given, all hits for the same query with scores exceeding the minimal score will be shown. Multiple hits are named <queryname>_(1), <queryname>_(2), etc. |
| --single_target_hits (or --chromosome) | prohibit Scipio to compose hits from multiple targets. Use this if you are sure that genes do not stretch out over multiple target sequences, for example if you search against full chromosomes. |
| Options controlling the output format | |
| --plusstrand = <value> | value for plus strand (default: '+') |
| --minusstrand = <value> | value for minus strand (default: '-'; some YAML parsers don't like this) |
| --verbal_strands | short for --plusstrand=forward --minusstrand=backward |
| --region_size = <value> | size of upstream/downstream regions (see keys "upstream"/"downstream" below; default: 1000 nucleotides) included in the output files |
| --show = <list> | which keys are to be shown in YAML output (details see below) |
| --show_intron = <list> | |
| --show_exon = <list> | |
| --show_gap = <list> | |
| --hide_undef | hide keys with undefined or empty list values |
| --hide_defaults | hide keys that have default values |
| Options passed to BLAT (ignored when BLAT is not run) | |
| --blat_bin = <name> | name of BLAT executable, defaults to "blat" |
| --blat_params = <params> | parameters passed to BLAT; see BLAT documentation. |
| --blat_tilesize = <..> | values for BLAT parameters -tileSize, -minScore and -minIdentity; by default, --blat_identity is set to 90% of --min_identity (e.g. the default --min_identity is 90, thus the default --blat_identity is 81). |
| --blat_score = <..> | |
| --blat_identity = <..> | |
| Parameters to adjust the Needleman-Wunsch algorithm (all penalty values are multiples of the mismatch penalty) | |
| --nw_insert_penalty = <value> | accounts for 3 missing nucleotides in the target sequence (there is an additional amino acid in the query sequence; default value is 1.5) |
| --nw_gap_penalty = <value> | accounts for 3 extra nucleotides in the target sequence (amino acid is missing in the query sequence; default: 0.8) |
| --nw_frameshift_penalty = <value> | accounts for 1 or 2 missing or additional nucleotides in the target sequence (default: 2.5) |
| --nw_intron_penalty = <value> | value for an intron of any size, with GT-AG pattern (default: 2.0) |
| --nw_stop_penalty = <value> | extra penalty for stop codons in the alignment (default: 2.5) |
| --exhaust_align_size = <value> | maximum sequence length for exhaustive search: because the Needleman-Wunsch algorithm is slow, the optimal alignment will not be computed in DNA regions longer than this (default: 500 nucleotides). Instead, Scipio will try here to place additional amino acids at the intron borders (resulting in one big intron), if possible, with few additional mismatches, or otherwise leave some amino acids unmatched (which will appear as "gap"). |
| --exhaust_gap_size = <value> | same as --exhaust_align_size, but referring to the query sequence instead of the target sequence (default: 3*Blat_tilesize). |
| Expert options | |
| --max_assemble_size = <value> | maximum size of intron parts at target boundaries. If an intron would have to be created between two partial hits across two contigs, that exceeds the given size (default: 75000 nucleotides), the two hits cannot appear together as parts of one composed hit. The contig with the lower score will be rejected (unless the score exceeds --min_score, and --multiple_results is enabled). |
| --min_dna_coverage = <value> | like --max_assemble_size, this option can be used to reject long introns joining partial hits; here, we require a minimal hit length/intron length ratio. Given in %, possible values are between 0 and 0.2%. (default: 0) |
| --min_intron_len = <value> | minimal length of an intron (default: 22 nucleotides). Any shorter sequence of extra nucleotides will be inserted into the exon as additional nucleotides in a sequence shift (see key "seqshift" below). |
| --transtable = <value> | let Scipio use an alternative genetic code table |
| --max_move_exon = <value> | this option determines how much Scipio shifts the BLAT prediction to find a correct intron (default: 2 amino acids). BLAT chooses between possible intron positions by minimizing mismatches. In rare cases, mostly cross-species alignments, there are several intron candidates causing an equal number of mismatches so that the correct one can only be recognized by matching the splice site consensus. In even rarer cases, the true intron location is more than two codons away from the BLAT prediction which is when you need this option. |
| --gap_to_close = <value> | maximum size of a gap in a query that Scipio closes by adding mismatches to exon boundaries (default: 6 amino acids). At gaps larger than that Scipio tries to insert additional exons. |
| --accepted_intron_penalty = <value> | GT---AG and GC---AG are by far the most common 3' and 5' splice sites, and thus by default excepted as correct intron borders. In very rare cases, other splice sites have been observed. To mark these introns as "intron" and not as "intron?" this parameter has to be given as 1.1 (to accept AT---AC intron borders) and 1.2 (to also except GG---AG and GA---AG intron borders). |
| Output overview | Back to top |
The script produces an output in YAML format. Additional scripts are supplied that transform the YAML output into other formats (e.g. GFF3). The output is organized as follows:
For each query, a list of the matched parts is given:
etc.
Usually, there is only one matched part (= BLAT hit) for each query, except in cases where queries are found on multiple non-overlapping targets (contigs).
At the end of the output, a list of the unmatched sequences is shown.
| Description of keys | Back to top |
Keys for BLAT hits
Every BLAT hit is described by the following keys:
| ID | The id of the BLAT hit (equals the line number in the psl file). |
| status | Might be "complete", "partial", "incomplete" or "manual". "complete" means that Scipio had no problems locating the query; "partial" means that the hit is on one of multiple targets each matching a part of the query; "incomplete" means that Scipio could not completely match the query sequence to the target; another reason for "incomplete" is a missing stop-codon in the target sequence following the last amino acid of the query sequence. "manual" can be entered if the output was modified by hand. |
| reason | If status is "incomplete", the reason why. |
| prot_len | The length of the query (in amino acid coordinates). |
| prot_start | Start of the matched part of the query if larger than zero. |
| prot_end | End of the matched part of the query if less than prot_len. |
| prot_seq | The query sequence (in a partial hit: only the matched part). |
| target | The name of the target sequence. |
| target_len | The total length of the target sequence. |
| strand | "+" (= forward) or "-" (= reverse). |
| dna_start | The location of the hit. |
| dna_end | |
| matches | The number of matches if less than prot_len. |
| mismatches | The number of mismatches. |
| undetermined | The number of undetermined residues. |
| unmatched | The number of unmatched query residues. |
| additional | The number of additional residues in the translated target. |
| score | The score of the hit. |
| upstream | DNA Sequence upstream of hit, ending before start codon. |
| upstream_gap | (unaligned) DNA Sequence preceding a partial hit. |
| matchings | The locations of exons and introns, see next section. |
| stopcodon | Stop codon if present. |
| downstream | DNA Sequence downstream of hit, starting with stop codon if present. |
| downstream_gap | (unaligned) DNA Sequence following a partial hit. |
The commandline parameter --show = <comma-separated list> can be used to choose a user-defined collection of keys to be shown.
For the alignment of the query to the translation, unmatched counts the gap characters inserted into the translation (= residues present only in the query), additional counts gap characters inserted into the query (= residues present only in the translation), matches+mismatches+undetermined counts residues present in both (see also comments to seqshifts below).
Keys for matchings
Every matching (intron/exon) is given with the following keys:
| type | "intron", "intron?", "exon", or "gap". "intron?" is used for uncertain introns (no regular splice patterns found) |
| nucl_start | Location in the query (in nucleotide coordinates; nucl_end not for introns). |
| nucl_end | |
| dna_start | Location in the target. |
| dna_end | |
| seq | DNA sequence of the feature. |
Keys that appear only in exons:
| seqshifts | A list of locations of sequence shifts. Each entry of the list consists in turn of four keys (nucl_start/end, dna_start/end). If nucl_start = nucl_end, the target sequence between dna_start < dna_end is translated and counted as additional residue(s); if dna_end - dna_start is not a multiple of 3, the extra single nucleotides causing the frameshift are translated as 'X'. If nucl_start < nucl_end and dna_start = dna_end, the corresponding part of the query sequence is missing in the translation and counted as unmatched. A frameshift caused my missing nucleotides is represented by a target segment of length 1 or 2 corresponding to one query residue (counted as undetermined). nucl_end - nucl_start is always a multiple of 3. |
| mismatchlist | Positions of mismatches (in amino acid coordinates). |
| undeterminedlist | Positions of undetermined residues (in query coordinates). |
| inframe_stopcodons | Positions of in-frame stopcodons (in query coordinates). |
| translation | Translation of the aligned part of the DNA sequence. |
| overlap | Number of nucleotides at the end of the target that are identical with the beginning of the following target, and not considered part of this location. |
| The commandline parameters --show_exon = <...>, --show_intron = <...>, --show_gap = <...> can be used to choose user-defined output for matchings. The following additional keys can be activated by this: | |
| nucl_pos | In introns, a synonym for nucl_start(=nucl_end) |
| prot_start | The location transformed into residue coordinates rather than nucleotides. A remainder of 1 is rounded down, and 2 is rounded up. |
| prot_end | |
| prot_pos | (this one for introns) |
| prot_seq | Part of the query matching an exon, or unmatched part in a gap. |
| Sequence coordinate conventions | Back to top |
All nucleotide and residue coordinates specify the number of preceding letters, that is, unlike in GFF, a location specified as starting at 5 and ending at 9 has length four, starting with the sixth and ending with the ninth character. (GFF would show this as start=6 end=9).
Locations on reverse strand are specified by negative numbers that represent the distance from the end of the reverse strand. Hence, a location specified as starting at -9 and ending at -5, refers to the reverse complement of the previous example (GFF would show this also as start=6 end=9).
Amino acid coordinates are rounded the following way:
If a location contains a split codon, our convention is to count it if its second nucleotide belongs to the location. Consequently, in reading frame 2 (meaning the last two nucleotides of the split codon are part of the next location) the start location is rounded down and the codon is considered part of the next location; conversely, a reading frame of 1 means that the codon is considered part of the previous location, and the starting point for the next location is rounded up when given in amino acid / codon coordinates.
| Overview of Scipio parameters as activity diagram | Back to top |
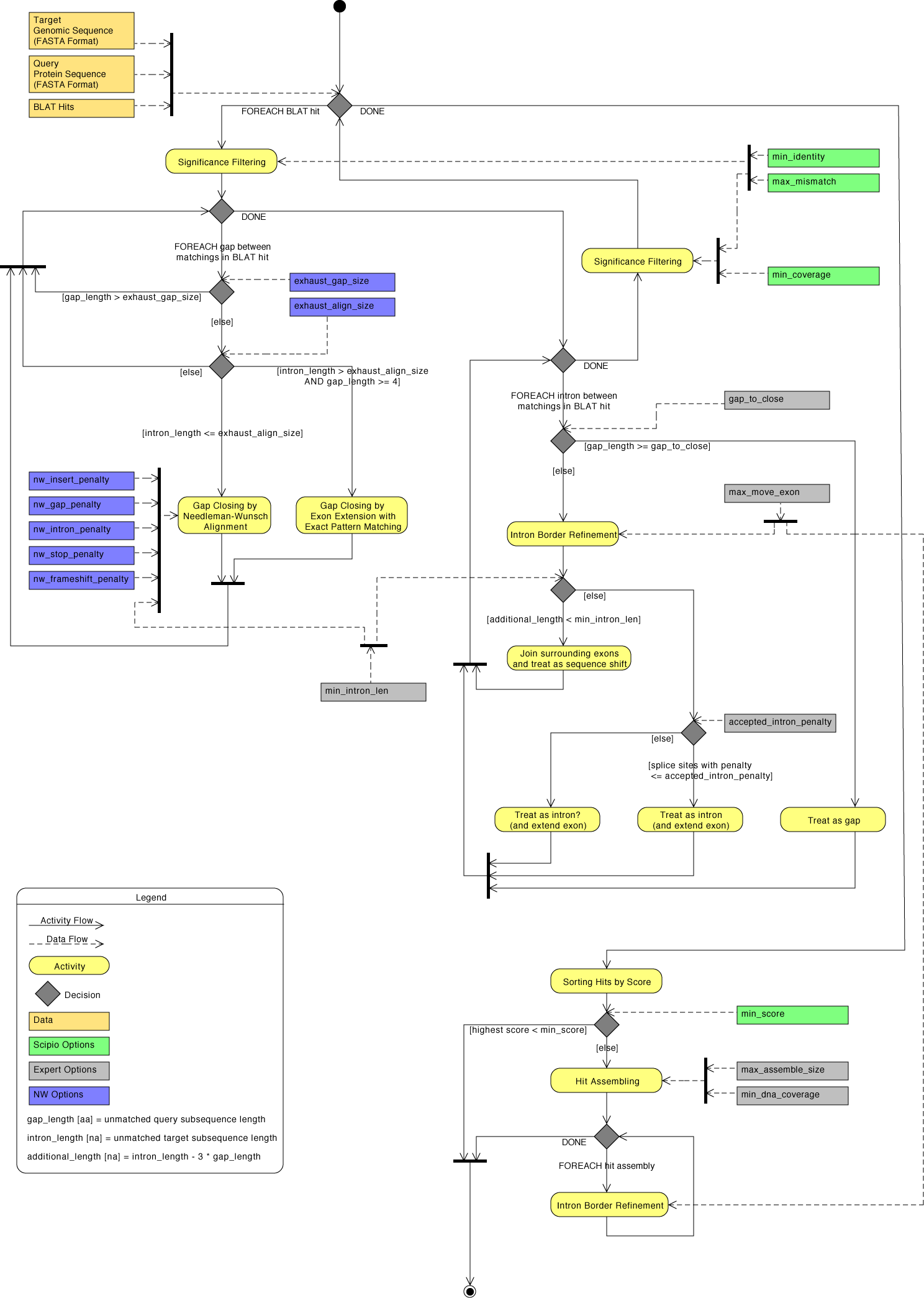
The general workflow of Scipio and WebScipio is as described in the publications and on the help pages. Scipio provides some general search parameters that filter the Blat ouput for further post-processing, and offers several expert options that influence the post-processing steps. In the new Scipio version, especially the part of the gap-closing (mapping the parts of the query sequence to the target sequence that Blat failed to recognize) and hit extension (modeling the regions at exon borders, including terminal exons, where homology was too low to be identified by Blat) has been improved (Figure). This has been done by implementing the Needleman-Wunsch algorithm for the search of unmapped query sequence in respective target regions and by introducing parameters that allow a higher divergence from the exon border regions predicted by Blat. All new parameters are adjustable by the user. The default values should be good enough for most cases, but especially when searching for very divergent homologs or when searching for homologs of very divergent species, these parameters need manual adaptation. Figure shows a detailed scheme of the Scipio workflow including all parameters that can manually be adjusted, and showing some of the most important decisions that Scipio makes to provide the best possible result. The detailed scheme should allow the experienced user to fine-tune the search in especially difficult cases.

|
| Detailed diagram of the Scipio application flow | Back to top |
|